During 2019, many AI healthcare companies announced FDA clearance for their medical device - this often considers AI radiology software. What does it actually mean when a company claims their artificial intelligence software is approved by the FDA? Why is this important? How does the FDA regulatory pathway for artificial intelligence radiology software currently work? And where do CADe and CADx versus AI fit in this story?
Why is it important that AI radiology software receives a nod by the FDA?
Well, for a start, if you want to use the software in clinical context, it is mandatory. However, as is often the case with rules, they exist for a reason. An FDA stamp of approval confirms the medical device in question has been extensively vetted by an authority that is specialized to do so. In other words, it provides certainty on the safety and efficiency of the device.
How does the FDA currently deal with AI radiology software?
The FDA has an extensive review process to determine whether a medical device is ready for clinical use. Which concrete steps the process contains is determined by what type of device is up for approval - a medical device, a device that does not claim to fulfill a medical purpose, or an investigational device.
Type 1: A medical device
Software that meets the definition of a medical device is regulated by the FDA. This means that software intended to be used for diagnosis, prevention, monitoring, treatment, or alleviation of a disease or injury may only be marketed for clinical use after it complies with FDA regulation. The FDA has set up a system in which devices, including software, have to be proven safe and effective for their intended use in various areas such as (datasets for) algorithm validation, design, (stand-alone and clinical) performance, usability, and also cyber-security. Furthermore, not only the products need to comply with FDA regulation, but also manufacturers themselves are required to operate according to the Quality System Regulations. This obligates manufacturers to have processes in place for controlled bug resolution, incident reporting, standardized design processes and overall risk management.
Type 2: The device does not claim to fulfill a medical purpose, e.g. research software
For software that does not fulfill a medical purpose, FDA regulations are not applicable. This means no evaluation of the clinical performance and no requirements on proper software design and cybersecurity measures; basically, no check that the device can fulfill its intended use safely and effectively. Such algorithms may be very suitable to use in a research setting and can offer additional features supporting research adequately, such as bulk export of data. However, they are not necessarily meant, nor designed to be as robust, safe, and effective as is required for clinical use (i.e. provide information that is used for individual patient management). Some hospitals, mainly academic ones, use such research software. This is usually developed in house and should be deployed for scientific research only. For example, software that provides volumetric data by automatically segmenting brain structures on MRI images can be a valuable addition to a research project.
Type 3: An investigational device
Software falls into the category of “investigational device” if it is part of a clinical trial to be evaluated on clinical performance with the purpose of obtaining FDA approval. This means the software is not cleared for support of clinical decision making just yet. Outcomes of the clinical trial first have to prove (or disprove, of course) that the device accurately provides the right information. However, performing a retrospective study, in which the software device is not directly used for clinical decision making, often offers a more straight-forward approach for obtaining regulatory clearance.1
An example of a study with an investigational device is stroke detection software that is integrated into the radiology workflow and used to diagnose patients, while other patients presenting with stroke related symptoms are diagnosed without the software. The stroke detection software will be part of a clinical trial of which the purpose may be to compare diagnosis decisions made with and without the software.
So where does AI fit in this story?
It might be a bit of an anti-climax, but whether a device belongs to any of the three categories discussed above does not have anything to do with whether the product is AI-based. A medical device can use artificial intelligence techniques, just like a “non-medical device” or an investigational device. But bear with us, we will get to the AI part in a bit.
The software classifies as a medical device. What’s next?
The FDA has determined over 1700 different generic types of devices and assigned those a product code and a classification as class I, II, or III. This classification is based on the risk the product poses and its intended use. The higher the risk of your product, the more stringent the process to get it FDA approved.
Class I and II
If a medical device searching clearance fits the criteria of an existing product code that is assigned to Class I or II, completing what is called the 510(k) program will provide FDA clearance for the device .2,3 The process is relatively straight forward: first, identify an FDA cleared device that is substantially equivalent to your device (this is called the predicate device), secondly, prove that the device performs at least at the same safety and effectiveness level. Again, there is no reason such a device cannot contain AI-technology. However, as many AI-techniques that are presently implemented in medical devices are relatively new and do not have an intended use that has been cleared in a previous device, chances are high that the device is not eligible for the 510(k) pathway. This immediately assigns the device to Class III.
Class II products are usually low patient risk, for example, powered wheelchairs, or devices with established techniques and materials that do not have life support functions, such as an X-ray mammography system or Ophthalmic system image management.
Class III
Class III devices usually sustain or support life, or present potential unreasonable risk of illness or injury to the patient. Undisputed devices in this category are all things implantable, such as pacemakers and stents. A device can be assigned to class III in two ways. First, when it fits one of the generic device groups as described by the FDA that already have a product code assigned and are determined to be class III. A second scenario is when the FDA has not coded and classified the device type yet. Then it is a new “device type” and there is no product code available. The FDA automatically assigns the device to class III. This is the case for many new healthcare concepts, including most AI-based software. This will require a device to go through the more extensive process of Premarket Approval (PMA) which, if successful, will lead to FDA approval prior to marketing your device. A PMA demands an applicant to provide “valid clinical information and scientific analysis”.4 In other words, clinical investigations, such as clinical trials, must be proved in addition to the technical and non-clinical studies which are already required for class I and II devices.
Not only is the FDA bill for the PMA process significantly higher than for the 510(k) process, but the required clinical trials are usually expensive as well as time consuming. Furthermore, extensive communication with the FDA can be expected throughout the review process, requiring a vast amount of resources and time to approval - significantly more than is necessary for a 510(k) process.
Thought to be class III, but found to be class II
For all those smaller companies out there that do not have the resources to cover the full course of a PMA process, there are two alternative approaches still open after being categorized in class III. Option 1: track down a predicate device. If you succeed at finding a medical device with the same intended use - which is substantially equivalent in safety and effectiveness - there is a big chance that product serves as a suitable predicate device for substantial equivalence determination. Meaning that the product is similar enough to the medical device up for approval to make it fit class II and therefore able to be cleared by completing a 510(k) process. Option 2 requires you to convince the FDA that the risks your device may put on patients is acceptable enough for the less stringent review pathway. This is done by sending in for review what is called a De Novo classification request.5 For this, you prepare technical documentation that contains a detailed description of the device, clinical and non-clinical performance tests, usability, and risk management activities. This is very similar to the way you prove safety and effectiveness in a 510(k) file. Furthermore, all the patient risks and patient benefits of the device have to be explained, including their mitigations, and propose special controls to ensure that the product is safe and effective in use. If the De Novo is granted, the device is categorized as Class II and the device group is now eligible for the 510(k) pathway with your device as a predicate device. If the FDA rejects your De Novo, there is no option left but to go for the more excessive PMA process.
The route of going through a De Novo process is actually a path that is often chosen by manufacturers seeking approval for their new AI-based devices.
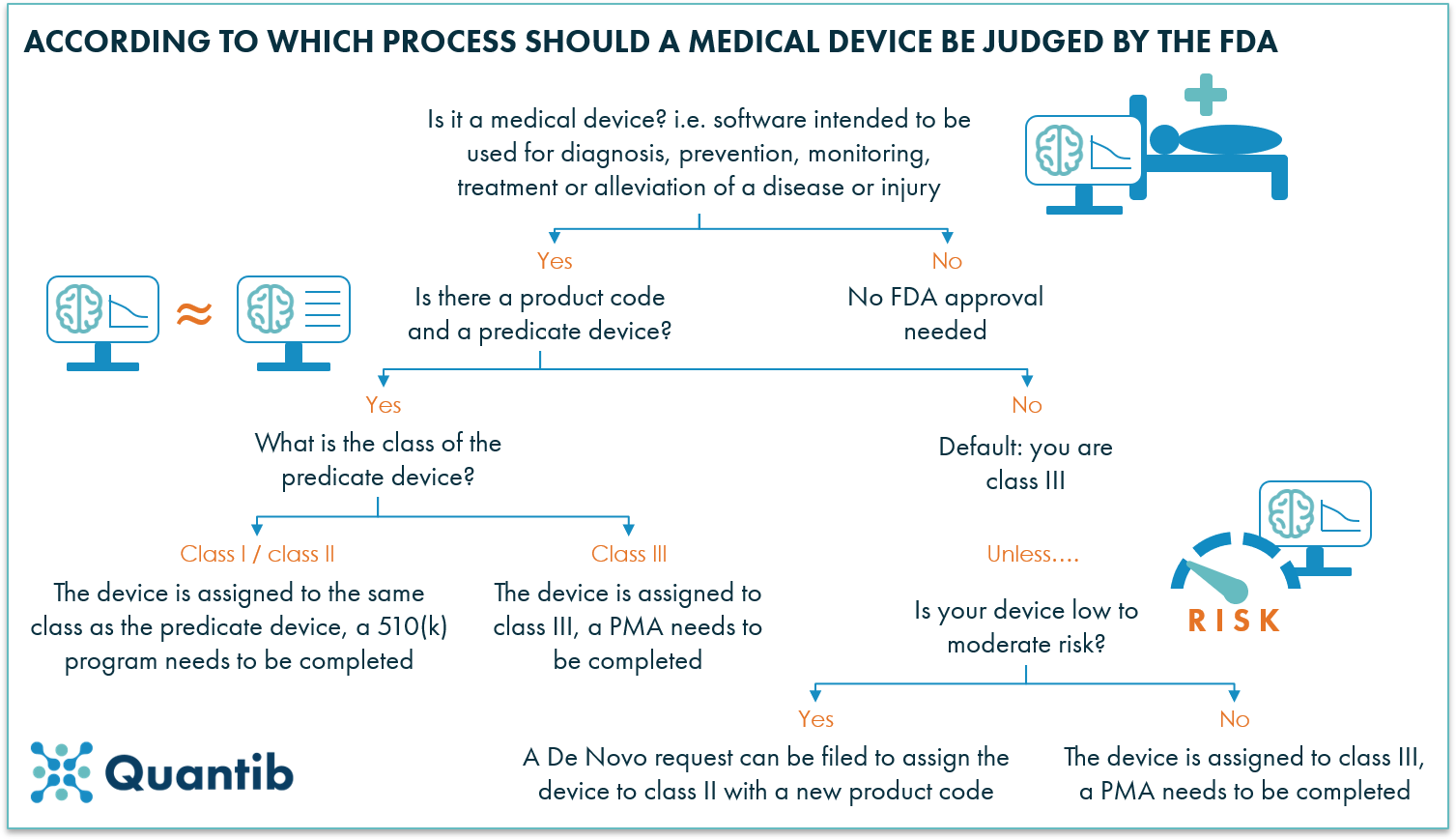
Figure 1: The FDA regulatory path consists of many processes. The figure above explains which process to follow, depending on the situation.

How about radiological CADe and CADx vs AI?
CADe and CADx, standing for Computer Aided Detection and Computer Aided Diagnosis, respectively, often create confusion when talking about AI in healthcare.
The definitions the FDA adheres to are as follows: a radiological CADe device is “intended to identify, mark, highlight or otherwise direct attention to portions of an image […] that may reveal abnormalities during interpretation of images by the clinician.” A CADx device is “intended to provide information beyond identifying […] abnormalities, such as an assessment of disease.” Whenever software is not intended to highlight an abnormality, it is not considered a CADe nor a CADx device. For example, segmentation of brain structures is not considered CADe, while the detection of a tumor candidate is. An algorithm that adds information on tumor grade would make it a CADx device.
So how does this relate to the different classes? In general, radiological CADx software will most likely fall under Class III at the time of writing, while radiological non-CADe software may find a Class II predicate device in the Image Analysis System (LLZ) product code.
Radiological CADe software can be both class II or class III, this is determined by the “regular process”. As soon as the device detects abnormalities, it is a CADe. Examples are CT lung nodule analysis and chest X-ray analysis. The category of CADe devices falling in Class II increases with every granted DeNovo. Lastly, there is a specific product code for devices that include borderline radiological CADe/CADx functionality. This is a wild card the FDA can play if the device is sort of CADe or CADx, but not really.
Still no word of AI. Non-CADe, CADe, and CADx software can all use artificial intelligence technology. CADe and CADx software tend to include AI more often, but a non-CADe device might just as well be deep learning-based. The FDA has published 2 specific guidance documents for evaluation of CADe devices. These documents provide direction on requirements for dataset collection, algorithm validation, setting up reader studies, and more - all very useful for AI radiology software.6,7
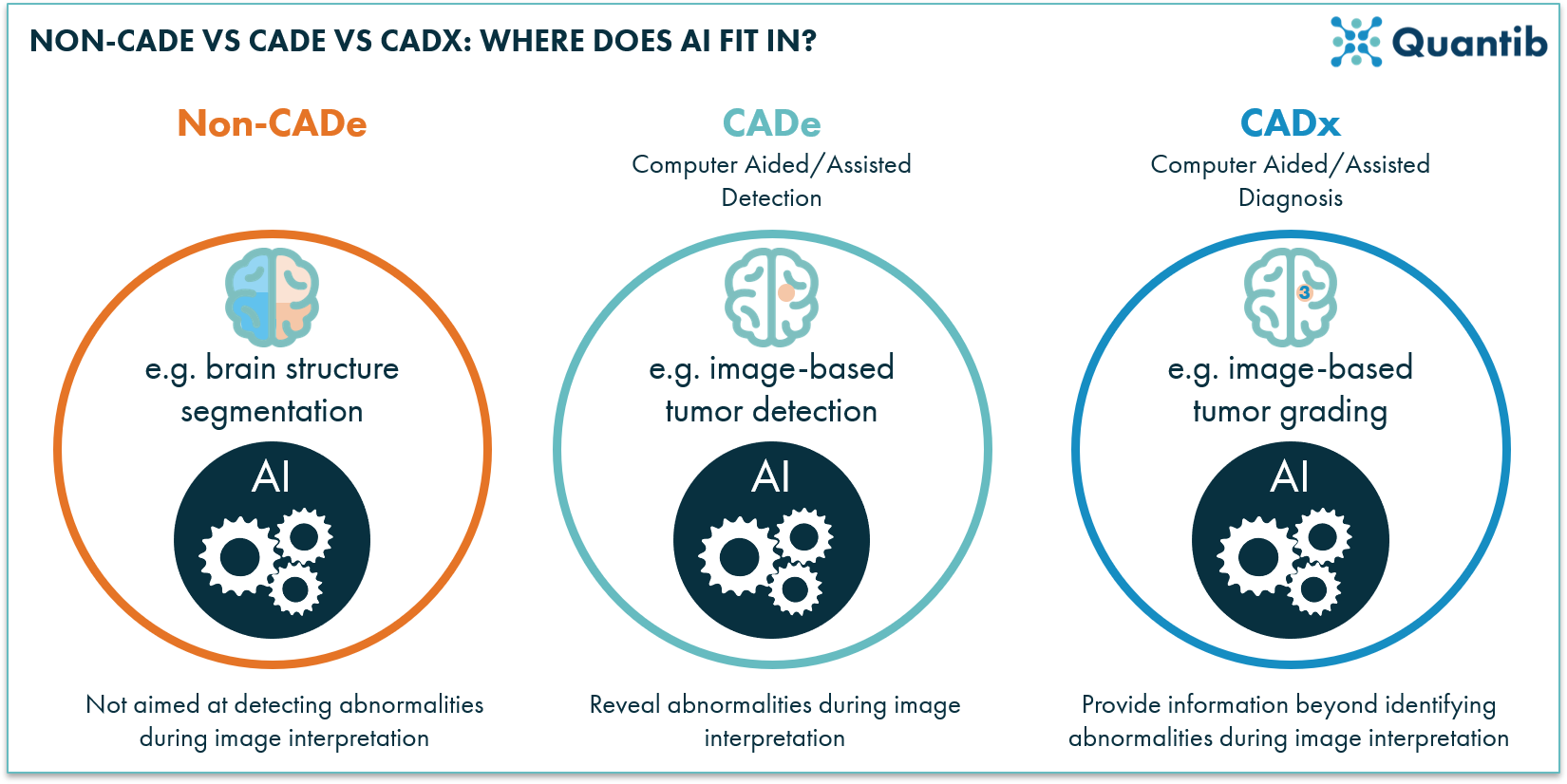
Figure 2: Non-CADe, CADe, and CADx - how do these types of products relate? All can be based on AI technology, but they have a different function.
So then what DOES the FDA say about AI?
Frankly speaking, not that much. Currently, the FDA works with guidelines that were developed in a pre-AI era. Most of the time, this is not a problem. As long as the manufacturer can provide detailed information on how the algorithm was developed and show that it is suitable for safe and effective use. In case of so-called continuous learning algorithms, i.e. algorithms that keep learning and therefore changing while being used in the clinic, the case becomes a bit more difficult. The FDA is working on guidelines covering this situation. To read more about this, check out our blog on how the FDA is planning to deal with continuous learning.
As a physician, what should you pay attention to?
Whether the FDA categorizes a device as a class II or III, or whether it is labeled CADe or CADx does not make a difference for how to use the device in the clinic. Even the presence of AI-based algorithms (or lack thereof) is not relevant. If you want to use a medical device in clinical practice, the only important factor is that it is cleared or approved to be used for the task you are planning to use it for.
Are you looking for software to support your research? Then non-approved algorithms might be able to do the job just fine. They are usually cheaper and easier to customize, which might be of benefit to the research. However, this is only possible because they are not tested for safety and effectiveness as extensively as software that is cleared or approved for clinical use. This is something to consider before making your pick.

Bibliography
- Device Advice: Investigational Device Exemption (IDE). Available at: https://www.fda.gov/medical-devices/how-study-and-market-your-device/device-advice-investigational-device-exemption-ide. (Accessed: 20th November 2019)
- The 510(k) Program: Evaluating Substantial Equivalence in Premarket Notifications [510(k)] | Guidance for Industry and Food and Drug Administration Staff. (2014).
- Premarket Notification 510(k). (2018). Available at: https://www.fda.gov/medical-devices/premarket-submissions/premarket-notification-510k. (Accessed: 20th November 2019)
- Premarket Approval (PMA). (2019). Available at: https://www.fda.gov/medical-devices/premarket-submissions/premarket-approval-pma. (Accessed: 20th November 2019)
- De Novo Classification Request. (2019). Available at: https://www.fda.gov/medical-devices/premarket-submissions/de-novo-classification-request. (Accessed: 20th November 2019)
- Computer-Assisted Detection Devices Applied to Radiology Images and Radiology Device Data - Premarket Notification [510(k)] Submissions - Guidance for Industry and Food and Drug Administration Staff. (2018). Available at: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/computer-assisted-detection-devices-applied-radiology-images-and-radiology-device-data-premarket . (Accessed: 20th November 2019)
- Clinical Performance Assessment: Considerations for Computer-Assisted Detection Devices Applied to Radiology Images and Radiology Device Data - Premarket Approval (PMA) and Premarket Notification [510(k)] Submissions - Guidance for Industry and FDA Staff. (2018). Available at: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/clinical-performance-assessment-considerations-computer-assisted-detection-devices-applied-radiology. (Accessed: 20th November 2019)